分子靶向治疗的功效可能受到肿瘤内同时发生的突变的限制。相反,这些改变可能会带来可以在治疗上利用的附带脆弱性。KRAS-突变肺癌的特点是肿瘤抑制基因的反复丧失STK11/LKB1.LKB1是否调节细胞对治疗应激的反应似乎是未知的。在这里我们展示了在LKB1-deficientKRAS-突变的肺癌细胞,KRAS或其下游效应物MEK的抑制由于NUAK介导的PP1B磷酸酶活性的丧失而导致JNK的过度活化。JNK介导的bcl-xl的抑制性磷酸化重新连接凋亡依赖性,使LKB1-deficient细胞易受MCL-1抑制。这些结果揭示了LKB1在调节应激信号和线粒体凋亡中的未知作用,而与AMPK和SIK介导的肿瘤抑制活性无关。此外,我们的研究揭示了LKB1-deficient中治疗诱导的脆弱性克拉斯-突变肺癌,可以作为基因型知情的策略,以提高KRAS靶向治疗的疗效。
导言KRAS的突变是一种调节MAPK/ERK信号传导的小GTPase,它定义了非小细胞肺癌 (NSCLC) 的最大遗传定义子集,占所有肺腺癌的25-30%
1。最近美国FDA和欧盟委员会批准的sotorasib (AMG 510)
2和adagrasib (MRTX849)
3,小分子共价KRASG12C-选择性抑制剂,标志着靶向治疗发展的一个里程碑
KRAS-突变的癌症.虽然大多数接受sotorasib治疗的NSCLC患者都获得了临床益处,但只有约40% 的患者获得了部分缓解
4。为了提高疗效,针对适应性耐药机制的药物联合策略
5,
6,
7,
8或者免疫逃避正在诊所测试。
KRAS-突变肺癌具有多种共同发生的突变
1尽管尚未完全表征,但新出现的证据表明,一些突变可能预示着对不同疗法缺乏反应。例如,
STK11/LKB1损耗和
KEAP1突变可能导致对包括抗PD-(L)1免疫检查点抑制剂在内的不同疗法缺乏反应
9,
10和KRASG12C抑制剂
3,
4。积极预测对KRAS的临床反应的同时发生的突变G12C抑制剂或药物组合仍未确定。考虑到遗传异质性
KRAS-突变肺癌,以及进入临床测试的多种药物组合,至关重要的是确定特定基因组改变所带来的脆弱性,并开发可以预测对KRAS反应的生物标志物G12C可能有助于指导患者选择的抑制剂组合。
临床前研究表明,敲低或抑制KRAS或下游信号传导
KRAS-突变细胞系通常不能诱导细胞凋亡
11,
12,
13。抑制MEK/ERK信号传导导致促凋亡BCL-2家族蛋白BIM的积累,这对于诱导细胞凋亡以响应一系列靶向治疗至关重要
14,
15。然而,由MEK或KRAS诱导BIMG12C单独的抑制往往不足以诱导细胞凋亡
KRAS-突变的癌细胞,因为BIM被促存活BCL-2家族蛋白如bclx-xl或MCL-1结合并中和。将MEK抑制剂与竞争性结合bcl-xl或MCL-1并释放BIM的BH3模拟物结合,可以诱导细胞凋亡并导致
KRAS-突变肿瘤
12,
16,
17。然而,临床相关的生物标志物可以区分特定的细胞凋亡依赖性 (MCL-1与bcl-xl),从而对患者进行KRAS治疗G12C缺乏抑制剂 BH3模拟组合。
在研究的反应
KRAS-KRAS的突变肺癌模型G12C或MEK抑制剂与BH3模拟物组合,我们出乎意料地观察到存在
STK11突变和对MCL-1的依赖。
STK11,它编码蛋白质LKB1 (肝激酶B1),在 ~ 30% 的
KRAS-突变肺癌
18。肿瘤抑制因子的丧失
STK11/LKB1
19通过调节能量平衡促进肿瘤发生
20,
21,增强转移潜能
22,
23,并启用免疫逃避
9,
24。然而,LKB1在调节细胞对治疗的反应中的作用在很大程度上未被探索。在这里,我们证明LKB1调节JNK应激信号和癌细胞的凋亡反应,而不依赖于AMPK介导的肿瘤抑制活性。
25,
26,
27和SIK
28,
29激酶。在LKB1-deficient
KRAS-用KRAS或MEK抑制剂处理的突变肺癌细胞,由于NUAK介导的PP1B磷酸酶活性的丧失,发生JNK的过度活化。JNK对bcl-xl的磷酸化导致对MCL-1的相互依赖性,使LKB1-deficient细胞易受MCL-1抑制。此外,我们的研究表明LKB1丢失是对KRAS敏感性的基因型标记。G12C-选择性抑制剂 + LKB1-deficient MCL-1抑制剂
克拉斯-突变肺癌细胞。
结果LKB1损失赋予对mapk MCL-1抑制的敏感性研究常见的共现基因组改变对KRAS的影响G12C针对不同途径的抑制剂组合策略,我们筛选了一组
KRASG12C-携带多种同时发生的突变NSCLC细胞系 (图。
S1A) 单独使用sotorasib或与靶向SHP2 (TNO 155),CDK4/6 (abemaciclib),PI3K (GDC-0941),bcl-xl/BCL-2 (navitoclax) 的抑制剂联合使用或MCL-1 (AMG 176) (图。
1A)。与先前对KRAS的研究一致G12C抑制剂
2,
7,
30,
31,我们观察到对KRAS的敏感性不同G12C抑制,这在我们有限的细胞系队列中是独立于最常见的同时发生的突变,如
TP53,
STK11/LKB1和
KEAP1(图。
S1B, C;补充数据
1)。一般来说,细胞系对其他抑制剂的总体敏感性较低,尽管少数细胞系对bcl-xl或MCL-1抑制表现出不同的敏感性 (图。
S1D)。这也与我们细胞系队列中同时发生的突变无关 (图。
S1E)。量化KRAS的功效G12C与KRAS相比的组合G12C单独计算,我们计算了AUC的相对变化 (e。g.,单一药物和组合剂量反应曲线之间的面积,标准化为单独索托拉西布的作用),下文简称为 Δ auc (图。
S2A)。正如预期的那样,在大多数细胞系中,sotorasib与其他抑制剂联合使用比单一药物sotorasib对细胞活力的抑制作用更大,尽管效果是可变的 (图。
S2B)。而同时发生的突变的存在似乎不影响靶向SHP2、CDK4/6或bcl-xl/BCL-2的组合的敏感性,具有同时发生的突变或
STK11/LKB1对靶向MCL-1或PI3K的组合更敏感 (图。
1B, C,无花果。
S2B)。PI3K抑制可以影响癌基因成瘾癌症中的多种细胞变化,包括mTOR依赖性MCL-1蛋白水平的下调
32,
33,我们证实了这一点 (图。
S2C)。为了进一步调查MCL-1在更大的队列中的作用
KRAS-突变的NSCLC细胞系,包括
KRAS除G12C以外的突变,我们测试了MEK抑制剂曲美替尼与AMG 176 (或相关化合物AM-8621
16)。同样,我们在LKB1丢失的细胞系中观察到trametinib amg 176的活性更高 (图。
1D,
S2D)。我们还用额外的MEK (cobimetinib) 和KRAS证实了这些发现G12C(阿达格拉西布) 抑制剂 (图。
S2E)。曲美替尼和AMG 176之间的协同活性导致联合活性的增加 (图。
S2F),导致组合产生净细胞毒性效应 (图。
S2G)。具有高 Δ auc值的LKB1-deficient细胞系在联合抑制KRAS/MAPK和MCL-1后表现出强劲的凋亡,而LKB1野生型 (WT) 细胞系的凋亡反应最小 (图
1E, F),表明LKB1可能调节细胞凋亡的依赖性
KRAS-突变肺癌.
A测试sotorasib药物组合的模式。B与单独的sotorasib相比,sotorasib + amg176组合的功效相对增加 (Δ auc-见图。
S2A用于解释) 反对
KRASG12C-突变的NSCLC细胞系。每个点代表一个独立的生物复制品,
N = 4)。Δ auc的比较
KRAS-突变的NSCLC细胞系分层根据
LKB1状态 *
p = 0.029 (C) 、 *
p = 0.032 (D),不成对-非参数
t测试,双面。
KRAS-突变的NSCLC细胞系用0.1 µ m的trametinib或1 µ m的sotorasib与1 µ m的AMG 176联合处理长达72 °h,并通过流式细胞术通过膜联蛋白阳性评估凋亡 (E,数据为平均值和S.E.M.
N = 3个生物学重复) 或活细胞成像 (F,数据为3次技术重复的平均值和S.E.M.)。对于膜联蛋白阳性,将媒介物组中凋亡细胞的百分比用作对照并归一化为0。源数据作为
源数据文件。
LKB1表达的调节改变细胞凋亡反应确定LKB1是否在调节细胞凋亡反应中起因果作用
KRAS-突变的NSCLC细胞,我们恢复了LKB1-deficient细胞系中的LKB1表达或删除了WT细胞系中的LKB1 (图。
S3A)。我们证实了通过增加经典LKB1激酶底物AMPK和ACC的磷酸化来恢复功能性LKB1活性 (图。
S3B,
S5B)。LKB1的重新表达降低了对sotorasib或trametinib MCL-1联合抑制的敏感性,相反,CRISPR介导的LKB1缺失使LKB1 WT细胞对sotorasib或trametinib + MCL-1抑制敏感 (图。
2A, B,
S3C, D)。LKB1的恢复或缺失不会改变对单独索托拉西布的反应 (图。
S3E) 或改变细胞增殖率 (图。
S3F),表明LKB1的获得或丧失时发生的对药物组合的敏感性变化主要是由细胞凋亡的MCL-1-dependent调节差异介导的。与这一观点一致,LKB1的恢复或删除分别减少或增加了sotorasib或trametinib amg 176的凋亡细胞死亡 (图
2C, D,
S3G),LKB1表达的恢复将细胞毒性反应转化为细胞抑制反应 (图。
S3H)。为了在体内证实这些结果,我们在小鼠中建立了等基因的H2030 EV和LKB1异种移植肿瘤。首先,与LKB1-null H2030 EV对照相比,我们通过增加LKB1和pAMPK染色证实了在已建立的异种移植肿瘤中LKB1功能活性的恢复 (图
S4A)。此外,我们测量了肿瘤中活性LBK1-SIK信号抑制的几种CRTC2/CREB靶基因的水平。
34,证实与LKB1-null对照相比,同基因LKB1-expressing异种移植肿瘤中的基因表达降低 (图。
S4B)。与体外结果相似,LKB1的恢复消除了H2030异种移植肿瘤对sotorasib或trametinib AMG 176的肿瘤消退 (图。
2E,
S4C)。总的来说,这些结果表明,LKB1的丢失使
KRAS-体外和体内对组合mapk + MCL-1抑制的突变NSCLC细胞。
A,B等基因LKB1-proficient和缺陷的相对 ∆ auc的比较
KRAS-突变细胞系对 (EV-空载体、LKB1-LKB1表达载体、sgGFP或GFP或LKB1的LKB1-CRISPR KO)。每个点代表一个独立的生物复制 (
N = 3-6)。对于Sotorasib,H2030: **
p = 0.002, H2122: *
p = 0.012,MGH1112-1: *
p = 0.014,MGH1114-1: *
p = 0.037, H358: **
p = 0.007。对于曲美替尼,2030年: **
p = 0.003, H2122: *
p = 0.026, H23: **
p = 0.002, A549: *
p = 0.007,MGH1114-1: *
p = 0.011,MGH1112-1: *
p = 0.015, H358: **
p = 0.0014, H441: ***
p = 0.0005, SW1573: **
p = 0.002。成对参数
t测试,双面。等基因的凋亡反应
KRAS-用0.1 µ m曲美替尼或1 µ µ m sotorasib与1 µ m AMG 176联合治疗后的突变NSCLC细胞系 (通过流式细胞术评估的膜联蛋白阳性 (C) 或活细胞成像 (D)。C每个点代表一个独立的生物复制品,
N = 3-5, H23: **
p = 0.003, MGH1112: *
p = 0.015, H2030: **
p = 0.0017,MGH9019-2: *
p = 0.011, SW1573: ****
p = 0.00001, H358: ****
p = 0.000015,不成对-非参数
t测试,双面。D数据为3次技术重复的平均值和s.e.M.。E从H2030 EV和H2030 LKB1细胞系建立皮下异种移植肿瘤,并用载体,sotorasib (每天30 μ mg/kg),trametinib (每天3 μ mg/kg),AMG 176 (每日50毫克/千克) 或组合。显示的数据是平均值和S.E.M
N = 每组5-6只小鼠,使用混合2-因素ANOVA效应模型确定单一药物和组合组之间的统计学差异,*
p = 0.01, **
p = 0.0084。源数据作为
源数据文件。
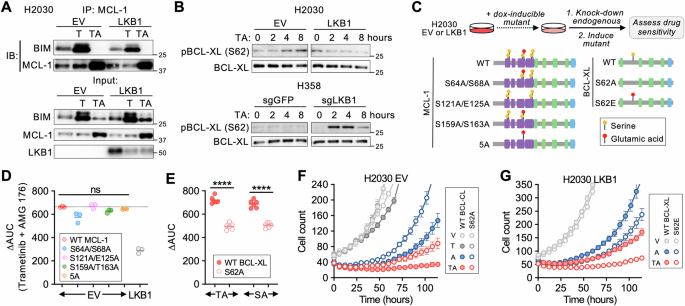
JNK激活与MCL-1依赖有关LKB1是一种主要的丝氨酸/苏氨酸激酶,调节多种细胞过程,包括生长
26,
35,细胞代谢
20,
21,和细胞极性
36,
37,
38。我们假设LKB1的丢失会重新连接下游激酶信号传导网络,从而赋予对MCL-1的依赖性,尤其是在致癌信号传导被破坏时。支持这一点,激酶死亡的LKB1的表达K781(kd) 突变体
25没有从mek + MCL-1联合抑制中拯救LKB1-deficient细胞 (图。
S5A, B),证明LKB1催化活性是观察到的药物敏感性差异所必需的。识别激酶信号的差异
KRAS-有或没有LKB1的突变NSCLC细胞,我们进行了基于质谱的全局磷酸化蛋白质组分析
39等基因H2030 (EV,LKB1和LKB1-kd) 和H358 (KO GFP,KO LKB1) 细胞在曲美替尼治疗前后 (图。
3A)。我们量化了27364种独特的磷 (图。
S5C、D),然后进行磷特征分析
40以鉴定在这些情况中的每一种情况下差异激活的激酶。与已知的MEK抑制对细胞周期进程的影响一致
41,我们观察到响应曲美替尼治疗的细胞周期相关磷酸化特征的下调,包括细胞周期蛋白依赖性激酶,ATM,ATR,极光激酶B和PLK1 (图。
S5E)。在没有药物治疗的情况下,LKB1野生型和缺陷细胞之间的激酶特征几乎没有统计学上的显著差异 (并且没有重叠) (图。
S5F),可能是营养丰富的细胞培养环境的结果。为了确定由LKB1调节的激酶活性的药物诱导的差异,我们寻找了相对于野生型对应物在曲美替尼处理的LKB1-deficient细胞中富集的激酶磷酸化特征 (H2030 EV对LKB1,H358 KO LKB1对KO GFP) 但未富集H2030 EV与激酶死亡的LKB1K87I细胞。虽然在曲美替尼处理的LKB1-deficient细胞中富集了两个等基因对,但只有一个特征-c-jun N端激酶1 (JNK1) -满足这些标准 (图。
3B)。具体来说,JNK1的成熟底物如ATF2、JUN和JUNB的磷酸化,与它们的LKB1野生型对相比,曲美替尼处理后在h2030ev和H358 k0 LKB1细胞中增加到更大程度。接下来,我们对trametinib + amg176处理后的H2030和H358等基因细胞进行了蛋白质组学分析。与对照细胞相比,H358 LKB1 KO细胞中的JNK磷酸化特征迅速增加 (8小时),并且JNK底物的子集在LKB1-deficient H2030细胞中显示出磷酸化增加 (图。
S5G, H)。这些结果表明,在曲美替尼或曲美替尼 + amg176组合抑制致癌信号传导后,LKB1丢失与JNK活化增加有关。
A等基因的磷酸化蛋白质组学分析
KRAS-用0.1 μ m曲美替尼处理48小时的突变NSCLC细胞系。B左: 曲美替尼处理的等基因细胞系对中磷酸肽特征的差异富集。NES-归一化富集分数。右,注释了JNK1下游底物的单个磷酸位点。C等基因H2030和H358细胞中响应mapk MCL-1抑制的磷酸-JNK变化 (数据为平均值和s.e.M.,每个点代表一个独立的生物复制,
N = 3-4, H2030: **
p = 0.0031, H358: ***
p = 0.0008, ***
p = 0.0009,成对参数
t测试,双面)。D具有siJNK1 + 2或阴性对照 (siNC) 的H2030 EV细胞的细胞数变化用0.1 µ m曲美替尼或1 µ m sotorasib与1 µ m AMG 176联合治疗后,通过活细胞成像定量。数据为3次技术重复的平均值和s.e.M.。E用靶向JNK1和2或siNC的sirna转染H2030 EV或LKB1细胞,然后用sotorasib (S) 或trametinib (T) ± AMG 176 (A) 处理并在3天后测定活力。每个点代表一个独立的生物复制 (
N = 3, TA: *
p = 0.017, SA: *
p = 0.028,不成对参数
t测试,双面)。FH2030 LKB1细胞中LKB1效应子的siRNA敲低示意图。G在不存在或存在amg176 (1 μ m) 的情况下,用sotorasib或曲美替尼处理用相应sirna转染的H2030 EV或LKB1细胞,并在3天后测定活力。每个点代表一个独立的生物复制 (
N = 3, ****
p = 0.00001,不成对参数
t测试,双面)。H,我用指定的sirna转染细胞,然后用曲美替尼 (0.1 µ m) 或sotorasib (1 µ m) 处理48 µ h,AMG 176处理4 µ h或曲美替尼 (0.1 µ m) 或索托拉西布 (1 µ m) 48 h,然后AMG 176 4 h。JsiPP1B恢复对组合索托拉西布或trametinib + AMG 176的敏感性 (∆ auc)。每个点代表一个独立的生物复制 (
N = 3, ****
p = 0.00001,双向方差分析)。KHA标记的wtnuak1 (WT) 或gkknuak在H2030等基因细胞中过表达,并且通过co-ip评估NUAK1和PP1B的相互作用。源数据作为
源数据文件。
为了证实这些结果,我们检查了H2030和H358等基因对中的JNK Thr183/Tyr185磷酸化。索托拉西布或曲美替尼 + AMG 176联合治疗导致H2030 EV细胞中JNK磷酸化的快速时间依赖性增加 (图。
3C,
S6A) 和JNK核易位 (图。
S6B)。JNK激活可以通过敲低MKK7来抑制,MKK7磷酸化并激活JNK (图。
S6C)。药物治疗后2小时即可观察到JNK激活,并在凋亡细胞死亡之前 (图。
S6D),与JNK激活在凋亡反应中的近端作用一致。LKB1的重新表达抑制了H2030细胞中的JNK磷酸化,相反,H358细胞中LKB1的缺失导致药物治疗后磷酸化JNK的增加 (图。
3C,
S6A)。我们通过比较一个更大的队列中磷酸化JNK的诱导来扩展这些发现
KRAS-用trametinib + amg176处理的突变NSCLC细胞。尽管细胞系之间存在预期程度的异质性,但与具有野生型LKB1的LKB1野生型细胞系相比,LKB1-deficient细胞系总体上显示出更大的JNK磷酸化诱导。pJNK诱导和组合敏感性之间存在显著相关性 (图。
S6E, F)。有趣的是,H1792细胞系在LKB1野生型细胞中表现出最大的药物敏感性 (图。
1B),显示出对pJNK的强劲诱导 (图。
S6E)。证实了H2030细胞的结果,H23细胞中LKB1的重新表达削弱了对trametinib amg 176的磷酸化JNK的诱导 (图。
S6G)。这些数据表明LKB1抑制在抑制致癌信号传导时发生的JNK依赖性应激信号传导。
由于JNKs调节细胞增殖,分化和存活以响应许多不同的环境和细胞应激物
42,我们检查了LKB1-deficient细胞中JNK信号的超激活是否对MAPK抑制具有特异性,还是反映了lkb1对JNK的调节作用。当H2030 EV或LKB1细胞暴露于紫外线时,一种成熟的JNK信号诱导剂
43,
44,我们观察到H2030 EV细胞中磷酸化JNK的增加在60 °min内达到峰值 (图。
S6H)。LKB1的重新表达减少了H2030 LKB1细胞中UV诱导的磷酸化JNK,表明LKB1可能在响应于各种刺激的抑制JNK应激信号中发挥一般作用。确定JNK激活是否是LKB1-deficient敏感性增加的基础
KRAS-突变的癌细胞结合mapk MCL-1抑制,我们使用siRNA同时敲低H2030细胞中的JNK1和2同工型 (图。
S6I),并评估对联合索托拉西布或trametinib + AMG 176的反应。虽然JNK1/2敲除对单独使用曲美替尼的敏感性几乎没有影响,但JNK1/2耗尽的细胞对两种药物组合的敏感性和凋亡反应降低。表型检测LKB1重新表达的影响 (图。
3D, E,
S6J)。我们将这些发现扩展到另外三个LKB1-deficient细胞系 (H23,H2122,LU65),其中JNK1/2的耗竭减少了对trametinib amg 176的凋亡反应 (图
S6K)。总的来说,这些结果表明,在缺乏LKB1的情况下,JNK信号的过度激活增加了LKB1-deficient的MCL-1依赖性。
KRAS-突变的NSCLC细胞,并使它们对组合的KRAS敏感G12C或mek + MCL-1抑制。
LKB1通过磷酸化和激活AMP激活的蛋白激酶 (AMPK) 家族的多个成员来发挥其作用。例如,LKB1通过感知增加的细胞内AMP/ATP比率和磷酸化AMPK在能量稳态中发挥核心作用,这反过来通过抑制mTOR和刺激自噬来抑制能量消耗
45。最近,AMPK相关的SIK激酶已被证明在介导LKB1对肿瘤发生和转移潜力的模型中发挥重要作用。
KRAS-突变型非小细胞肺癌
28,
29。然而,LKB1在调节细胞凋亡引发中的作用在很大程度上是不确定的。为了鉴定介导LKB1对药物诱导的JNK激活和MCL-1依赖性的抑制作用的LKB1底物激酶 (s),我们同时沉默了在NSCLC中表达的每个AMPK相关激酶家族中的多个成员的表达。
29(图。
3F,
S7A-D)。沉默NUAK1 2足以将H2030细胞对组合的sotorasib或trametinib amg 176的敏感性恢复到与H2030细胞LKB1-deficient相似的水平 (图。
3G,
S7E)。相比之下,在LKB1重新表达的背景下沉默SIKs、AMPKs或MARKs并不能恢复药物敏感性 (图。
3G,
S7F)。此外,当细胞在高或低/无葡萄糖条件下培养时,LKB1-deficient和LKB1-restored细胞之间的药物敏感性差异相似 (图
S7G),符合营养独立机制。我们验证了NUAKs的敲低,而不是其他LKB1底物的敲低,恢复了表达LKB1的H23和MGH1112-1细胞的药物敏感性 (图。
S7H)。在表达LKB1的H2030、H23、MGH1112-1细胞中,NUAK1/2的敲除恢复了药物诱导的JNK磷酸化,其水平与其匹配的等基因对照相似 (图。
3H,
S8A, B),并增加LKB1-expressing细胞对trametinib amg 176的凋亡反应 (图。
S8C)。
NUAKs调节细胞极性
46,倍性
47,和附着力
48通过肌球蛋白磷酸酶靶向-1-蛋白phosphatase-1beta (PP1B) 复合物的磷酸化。NUAK1通过置换自抑制蛋白I-2直接结合并激活PP1B磷酸酶
48。我们假设LKB1-NUAK1下游的PP1B激活可能导致JNK的去磷酸化。PP1B表达的敲低显著增加了LKB1-restored H2030细胞中的pJNK (图。
3I) 和对mapk MCL-1抑制的敏感性增加 (图。
3J,无花果。
S8D),表明PP1B使JNK去磷酸化并降低lkb1下游的MCL-1依赖性。演示NUAK1是否在LKB1-expressing中直接与PP1B相互作用
KRAS-突变的NSCLC细胞,我们在H2030、H23、MGH1112-1 EV和LKB1细胞中表达HA标记的NUAK1。PP1B的共免疫沉淀 (co-ip) 显示H2030 LKB1细胞中NUAK与PP1B的结合增加 (图。
3K,比较泳道1和3),其被先前已证明介导NUAK-PP1B相互作用的NUAK GILK结构域 (GKKK) 的突变破坏
48(图。
3K,比较车道3和5)。相反,I2蛋白与PP1B的结合在h2030lkb1细胞中减少,而在NUAK GKKK突变体存在下增加,这与NUAK和I2之间结合PP1B的竞争LKB1-dependent一致。这些结果在H23和MGH1112-1细胞中得到概括,与EV对照相比,LKB1表达细胞中NUAK1和PP1B之间的相互作用增加 (图。
S8E, F)。总的来说,这些结果表明,由于PP1B磷酸酶活性降低,LKB1-NUAK1/2信号传导的丧失导致JNK信号传导增加,从而导致对组合的mapk MCL-1抑制的敏感性增加。
抑制MEK/ERK信号传导导致BIM积累并增加用各种靶向疗法治疗的癌基因驱动的癌症中的细胞凋亡引发,驱动细胞进入MCL-1和/或bcl-xl依赖状态
15,
17。为了证实LKB1调节凋亡引发,我们进行了BH3分析
49,
50,
51在用曲美替尼治疗前后的等基因LKB1-deficient或WT细胞系上 (图。
S9A)。正如预期的那样,曲美替尼治疗增加了总体凋亡引发 (图。
S9B)。与LKB1野生型细胞相比,曲美替尼在LKB1-deficient中诱导更大的MCL-1特异性引发 (表示为 “Δ 引发”) 增加,并且在LKB1重新表达后持续降低 (图。
S9C, D)。相反,H358细胞中LKB1的缺失增加了曲美替尼诱导的MCL-1依赖性。在一组细胞系中,我们还观察到bcl-xl依赖性的变化,但这不是一致的效果 (图。
S9E)。为了研究LKB1-deficient细胞中MCL-1依赖性引发增加的基础,我们检查了MCL-1蛋白表达水平,因为这高度依赖于由mTOR调节的帽依赖性翻译。
52(由AMPK监管)。LKB1、MCL-1和bcl-xl蛋白表达在LKB1-deficient和野生型中相似,与AMPK非依赖性作用一致。
KRAS-突变的NSCLC细胞系 (图。
S9F, G) 或等基因细胞系对 (例如,参见图。
S10E)。接下来,我们检查了BIM和MCL-1或bcl-xl之间的相互作用。Co-ip实验显示曲美替尼治疗后与MCL-1和bcl-xl结合的BIM增加 (图。
S10A-C),与先前的研究一致
16。与LKB1野生型细胞系相比,用曲美替尼处理的LKB1-deficient细胞与MCL-1结合的BIM更多,与bcl-xl结合的BIM更少 (图。
S10A-C)。在曲美替尼治疗后,缺陷细胞系中LKB1的恢复减少了与MCL-1结合的BIM的量,而在野生型细胞中敲除LKB1增加了与MCL-1结合的BIM的量 (图。
4A,
S10D-G)。值得注意的是,除了一种细胞系 (A427) 之外,在不存在药物处理的情况下,LKB1再表达/敲低对基线BIM:MCL-1结合的影响较不显著。这些结果表明,LKB1的丧失促进BIM:MCL-1复合物的形成,特别是在抑制致癌MAPK信号传导,功能性诱导MCL-1依赖状态和引发amg176敏感性的情况下。
A用载体,曲美替尼 (0.1 µ m) 处理后,BIM与H2030 EV (空载体) 和H2030 LKB1细胞中的MCL-1结合的co-ip对于24 h或曲美替尼24.H,然后是AMG 176 (1 µ m) 4.H。B用0.1 µ m trametinib 1 µ m AMG 176处理后,通过蛋白质印迹在等基因H2030和H358细胞中bcl-xl S62磷酸化的时程。C表达MCL-1和bcl-xl磷酸位点突变体同时抑制内源MCL-1和bcl-xl的实验方法。询问的磷酸化位点以黄色表示,磷酸模拟位点以红色表示。DMCL-1磷酸化位点突变体不会降低对MCL-1抑制的敏感性 (Δ auc)。在诱导突变MCL-1 (或WT对照) 和敲低内源性MCL-1后,在不存在或存在AMG 176 (1 µ m) 的情况下用曲美替尼处理H2030 EV细胞并在3天后测定活力。每个点是一个独立的生物复制 (
N = 3)。EBcl-xls62a突变体降低了MCL-1依赖性。诱导bcl-xl S62A (或WT对照) 和敲低内源性bcl-xl后,用单独的sotorasib或trametinib或在amg176 (1 μ m) 存在下处理H2030 EV细胞,并在3天后测定活力。每个点是一个独立的生物复制 (
N = 6, ****
p = 0.000001,不成对参数
t测试,双面)。表达诱导型WT或S62A突变体bcl-xl S62A的H2030 EV细胞 (F) 或表达诱导型WT或bcl-xl S62E磷酸模拟物的H2030 LKB1细胞 (G) 用0.1 µ m曲美替尼或0.1 µ m曲美替尼与1 µ µ m AMG 176联合处理,并通过活细胞成像定量细胞数。数据为3次技术重复的平均值和s.e.M.。V车辆,T trametinib,A AMG 176,TA trametinib + AMG 176。源数据作为
源数据文件。蛋白质印迹和免疫沉淀图像代表至少2个独立的生物学重复。
MCL-1和bcl-xl可以被许多激酶 (包括JNK和ERK) 在多个残基处磷酸化,从而导致对蛋白质稳定性/降解,BIM结合亲和力和细胞凋亡的上下文特异性和不同影响
53,
54,
55,
56,
57,
58。曲美替尼治疗后,T163处的MCL-1磷酸化急剧下降,与ERK磷酸化丧失一致
59然后在稍后的时间点反弹,与JNK的激活一致 (图。
S11A)。LKB1-deficient细胞中LKB1的恢复减少了MCL-1磷酸化的反弹,而野生型细胞中LKB1的缺失增加了MCL-1磷酸化 (图。
S11A, B)。在LKB1-deficient细胞中观察到类似的时间和JNK依赖性模式的bcl-xl在S62处的磷酸化,其被lkbl的再表达抑制。在用trametinib amg 176的组合处理后,bcl-xl S62在LKB1-deficient但未LKB1-proficient的同基因细胞系对中被迅速磷酸化 (图
4B)。沉默JNK1/2表达将药物诱导的MCL-1和bcl-xl的磷酸化降低到与相应的LKB1-restored等基因细胞系相似的水平 (图。
S11C,比较车道3、4和7)。评估JNK介导的MCL-1或bcl-xl磷酸化是否影响药物敏感性,我们在H2030细胞中表达了DOX诱导型MCL-1或bcl-xl磷酸化位点突变体,同时敲低了内源性MCL-1或bcl-xl的表达 (图。
4C,
S11D-G)。虽然将MCL-1磷酸化位点突变为丙氨酸对trametinib amg 176的敏感性影响很小 (图。
4D,
S11H),bcl-xl S62A突变体的表达降低了H2030和其他细胞系对sotorasib或trametinib amg 176的敏感性 (图
4E, F,
S11K) 、表型选择LKB1重新表达和JNK1/2敲低。相反,bcl-xl S62E磷酸模拟物增加了H2030 LKB1细胞的敏感性 (图。
4G)。这些结果表明,LKB1-deficient细胞的MCL-1依赖性增加是由bcl-xl磷酸化介导的。
先前的研究表明,癌细胞对MCL-1抑制的敏感性与bcl-xl表达水平和bcl-xl中和促凋亡BH3蛋白如BIM的能力成反比。
60,
61。Bcl-xl S62的磷酸化诱导构象变化,其中失调的结构域折叠到bcl-xl BH3结合槽中,以防止BIM结合
58。因此,我们假设JNK对bcl-xls62的磷酸化损害了bcl-xl螯合MCL-1抑制后从MCL-1释放的BIM的能力。为了测试这一点,我们研究了BIM:MCL-1和BIM: bcl-xl相互作用的动力学,首先用曲美替尼处理细胞以增加与MCL-1结合的BIM,然后用AMG 176的短脉冲治疗,并评估bcl-xl隔离从MCL-1释放的BIM的能力 (图。
5A)。在LKB1-deficient H2030细胞中,与LKB1野生型SW1573细胞相比,在用AMG 176处理后,bcl-xl隔离的BIM非常少。它表现出bcl-xl对BIM的大量隔离 (图。
5B)。在H2030细胞中恢复LKB1表达或沉默JNK1/2增加了加入AMG 176后bcl-xl隔离的BIM量 (图。
5C, D)。在H2030和MGH1112-1 EV细胞中,bcl-xl S62A突变体表现出增加的BIM: bcl-xl结合,而在H2030 LKB1细胞中,磷酸模拟S62E突变体降低了BIM: bcl-xl结合 (图。
5E, F,
S11L)。H2030细胞中NUAK1/2表达的敲低,我们显示恢复了药物诱导的JNK磷酸化 (图。
3H),恢复了药物诱导的bcl-xl S62磷酸化 (图。
5G)。总的来说,这些结果表明,在LKB1丢失的情况下,JNK的激活通过磷酸化bcl-xl并降低其缓冲BIM促凋亡作用的能力来产生MCL-1依赖性状态 (图。
5小时)。虽然在某些情况下,特别是那些可能是高度引发和MCL-1依赖在基线,LKB1的损失可能赋予MCL-1抑制单独的敏感性,MCL-1依赖性通过抑制致癌MAPK信号传导后凋亡引发的增加而增强。
A从MCL-1置换后调查BIM封存的方法方案。BH2030 (LKB1-deficient) 和SW1573 (LKB1野生型) 中与MCL-1和bcl-xl结合的BIM的co-ip评估用0.1 μ m曲美替尼处理24小时,然后用1 μ m AMG 176处理4小时后的细胞。CCo-ip评估与bcl-xl结合的BIM,并在用0.1 µ m曲美替尼处理24 °h,然后用1 µ m AMG 176处理4 °h后在H2030 EV和LKB1细胞中MCL-1。D绑定到bcl-xl并在H2030 EV中MCL-1的BIM的co-ip评估 (空向量) 用0.1 µ m曲美替尼治疗24 h +.1 µ m AMG 176 4 h后,JNK敲除。E在H2030 EV (S62A) 和H2030 LKB1 (S62E) 中与WT bcl-xl或bcl-xl突变体结合的BIM的co-ip评估用0.1 μ m曲美替尼处理24小时后的细胞,然后用1 μ m AMG 176处理4小时。HA-标签下拉对于可诱导的构建体是特异性的。F在过表达bcl-xlwt或S62A突变体的H2030和MGH1112细胞中与bcl-xl结合的BIM的co-ip评估的定量。数据为平均值和s.e.M.,每个点代表一个生物复制品 (
N = 3-5, H2030: *
p = 0.0433,MGH1112-1: *
p = 0.0345,不成对参数
t测试,双面)。G0.1 µ m曲美替尼治疗48 h (T) 或曲美替尼治疗48 h后1 µ m AMG 176 (TA),NUAK1/2敲低对bcl-xl S62磷酸化的影响4 h。H描述LKB1丢失导致MCL-1-dependent状态并使其敏感的机制的模型
KRAS-联合KRAS或mek + MCL-1抑制的突变nsclc。源数据作为
源数据文件。蛋白质印迹和免疫沉淀图像代表至少2个独立的生物学重复。
LKB1-deficient患者和PDX肿瘤依赖MCL-1为了研究我们发现的临床相关性,我们对
KRAS-离体暴露于索托拉西布或曲美替尼后的突变nsclc (从患者恶性胸腔积液中分离出的实体转移性病变或肿瘤细胞) (图。
6A)。sotorasib和trametinib治疗均导致MCL-1依赖性启动 (MS1肽) 增加
STK11/LKB1-突变体,但不是WT肿瘤,(图。
6B,
S12A)。与这种效果一致,对从同一患者获得的恶性胸腔积液中分离的肿瘤细胞进行的免疫共沉淀实验显示,药物诱导的与MCL-1结合的BIM增加 (图。
6C)。相比之下,我们没有观察到药物诱导的bcl-xl依赖性引发 (HRK肽) 在
STK11-突变体和WT肿瘤。为了扩展这些发现,我们对
KRAS-突变 (G12C和其他) NSCLC患者来源的异种移植 (PDX) 模型,有或没有共同发生
STK11曲美替尼短期治疗后的损失。与患者肿瘤和体外细胞系模型相似,与WT肿瘤相比,LKB1-deficient肿瘤表现出增加的MCL-1-dependent引发 (图。
6D,
S12B)。在LKB1-deficient PDX肿瘤中,将AMG 176添加到sotorasib中导致的肿瘤反应比单独使用sotorasib更大,但LKB1-deficient PDX肿瘤没有MCL-1-dependent引发 (图。
6E, F,
S12C、D)。LKB1-deficient PDX肿瘤在sotorasib + AMG176治疗后表现出明显的JNK磷酸化,这在LKB1野生型PDX肿瘤中未观察到 (图。
S13A)。我们证实,在药物治疗的LKB1-deficient,而不是LKB1-expressing,等基因的H2030异种移植肿瘤中,磷酸化-JNK有类似的增加 (图。
S13B)。为了研究潜在的毒性,我们评估了间歇性AMG 176给药的联合给药方案 (在目前正在进行的临床试验中,AMG 176作为间歇性输注给药),该方案诱导了类似的肿瘤消退 (图。
S13C)。在人源化MCL-1敲入小鼠中
61sotorasib与AMG 176的组合耐受性良好,没有明显的毒性迹象 (图。
S13D)。与目标MCL-1抑制的预期效果一致
61,我们观察到b细胞和单核细胞减少,但是与单独的AMG 176相比,与sotorasib组合没有观察到额外的作用 (图。
S13E)。因此,LKB1肿瘤抑制因子的丧失与MCL-1对索托拉西布或曲美替尼治疗的依赖性增加有关。
KRASG12C-突变的nsclc,产生可通过同时抑制MCL-1来利用的凋亡脆弱性。
A
KRASG12C-在用sotorasib或trametinib离体处理后,收集突变的NSCLC肿瘤细胞用于BH3分析和BIM:MCL-1相互作用的评估。BMCL-1 (MS1 10 + 30 µ m肽) 和bcl-xl (HRK 10 + 100 µ m肽) 的变化用0.1 μ m曲美替尼或1 μ m sotorasib治疗离体治疗后患者肿瘤细胞的依赖性引发。数据标准化为车辆控制,误差条显示S.E.M. (
N = 3,6. *
p = 0.0476,不成对-非参数
t测试,双面)。CBIM的co-ip评估: 用0.1 μ m trametinib (T) 或1 μ m sotorasib (S) 离体处理16小时后,从胸膜液中分离出的肿瘤细胞中的MCL-1相互作用。数据代表2个独立的生物学重复。D小鼠轴承
KRASG12C-将突变型NSCLC患者来源的异种移植物 (PDX) 肿瘤用sotorasib (100 μ mg/kg) 处理3天并收获用于BH3分析。显示的数据是标准化为载体对照的sotorasib处理的肿瘤的MCL-1依赖性引发 (MS1肽) 的差异,每个点代表独立的小鼠肿瘤,误差条为s.e.M. (
N = 3-7, ***
p = 0.0002,双因素方差分析)。E小鼠轴承
KRASG12C-用媒介物sotorasib (100 °mg/kg) 治疗突变型NSCLC PDX肿瘤 (LKB1-loss: MGH1112-1,MGH1138-1,MGH1196-2; LKB1 WT: MGH1062-1,MGH1145-1,MGH10199-3)。每日AMG 176 (50 mg/kg) 或sotorasib (100.Mg/kg) + AMG 176 (50.Mg/kg)。显示的数据是平均值和S.E.M.
N = 每组6-10只动物,使用混合效应模型确定单剂和组合组之间的统计学差异,****
p = 0.00001, *
p = 0.013,双因素方差分析)。FPDX荷瘤小鼠的生存曲线: 无进展生存期 (PFS) 指标由从基线测量到肿瘤体积增加20% 的时间确定 (****
p = 0.0001, **
p = 0.0087,对数秩Mantel-Cox检验)。源数据作为
源数据文件。A-D在BioRender中创建。李,C。(2025年)
https://BioRender.com/p50l332。
讨论虽然靶向肺癌中的躯干致癌驱动突变的效用是牢固确立的,但大多数临床靶向治疗策略不考虑同时发生的突变。用于
KRAS-特别是突变型肺癌,确定与肿瘤抑制基因中反复出现的同时发生的突变相关的脆弱性,可以开发生物标志物驱动的联合疗法,在不同的患者亚群中具有增强的活性。然而,目前在临床上的大多数KRAS抑制剂药物组合的开发对于同时发生的突变是不可知的。我们的发现,LKB1调节凋亡依赖性
KRAS-突变肺癌是出乎意料的,因为与癌基因成瘾的实体瘤中对BH3模拟物敏感性相关的基因组特征一直难以捉摸
12,
16,
17。失活突变或丧失
STK11/LKB1,它定义了一个主要的基因组亚组
KRAS-突变肺癌
1,
19,
62,特别令人感兴趣,因为它们与对免疫检查点阻断的反应性降低有关
9,
63总体预后差
64。
LKB1是一种主激酶,通过磷酸化AMPK家族激酶的多个成员来调节多种细胞过程。
45,
65。特别地,LKB1在通过AMPK调节能量稳态中的作用已经被很好地定义。在能量压力 (高AMP:ATP比率) 的情况下,AMPK通过TSC2抑制mTORC1来限制合成代谢过程
66。有趣的是,MCL-1的表达水平高度依赖于mTOR介导的帽依赖性翻译,并且小分子抑制剂对mTOR的抑制已被证明可以减少MCL-1表达并赋予凋亡敏感性。
33。我们还观察到LKB1-deficient中PI3K抑制,MCL-1下调与AMG 176敏感性之间的关联。
KRAS-突变的NSCLC细胞系。然而,我们没有观察到操作LKB1后MCL-1表达的任何变化,并且沉默AMPK表达并没有表型化LKB1丧失对MCL-1抑制剂敏感性的影响。总的来说,这些结果支持独立于AMPK的机制,LKB1通过该机制调节JNK信号传导和MCL-1依赖性。
除了通过AMPK调节代谢的作用外,LKB1丢失还通过重新编程表观遗传状态,促进谱系可塑性和促进转移来促进肿瘤发生。
22,
67,
68,
69,
70。最近的研究揭示了AMPK相关的SIK激酶在介导LKB1对肿瘤发生的抑制作用中的核心作用,对NUAKs具有潜在的促进作用
28,
29。NUAK激酶已被证明可以调节正常组织中的细胞极性,粘附和细胞周期
46,
48,
71并在神经突形成中发挥关键作用
72。我们的结果表明,NUAKs可以通过结合和激活JNK磷酸酶PP1B来充当JNK信号传导的负调节剂。据我们所知,LKB1/NUAK1/PP1B轴代表了LKB1可以抑制JNK应激信号传导并调节细胞凋亡的机制。据报道,JNK通过磷酸化多个促凋亡和抗凋亡BCL-2家族成员来调节凋亡信号传导,包括BIM
73,
74,
75,
76、BAX
77,
78,
79,BCL-XL
56,
57和MCL-1
53,
55,
80,
81。差异磷酸化的后果是复杂的,并且可以影响蛋白质稳定性/周转以及蛋白质-蛋白质相互作用,从而以上下文特定的方式导致促凋亡和抗凋亡作用。我们观察到JNK介导的MCL-1和bcl-xl对KRAS和MEK抑制的反应,然而,bcl-xl中JNK磷酸化位点的消除,而不是MCL-1的消除,证实了用JNK敲低或LKB1再表达观察到的MCL-1依赖性的降低。未来的研究将有必要确定MCL-1的JNK磷酸化是否可能在其他治疗背景下赋予细胞凋亡脆弱性。有趣的是,我们观察到LKB1-deficient细胞系的子集在不存在MAPK抑制的情况下表现出对单一试剂MCL-1抑制的敏感性,表明高度引发的MCL-1-dependent基线状态。LKB1的重新表达部分降低了对MCL-1抑制的敏感性,这表明在某些情况下,LKB1/NUAK对JNK的基线抑制可能会影响在没有治疗应激的情况下的凋亡依赖性。通过在抑制致癌MAPK信号传导的情况下发生的增加的凋亡引发进一步放大。
虽然我们的研究集中在KRAS-用KRAS或MEK抑制剂靶向疗法治疗的突变型肺癌,我们还提供了LKB1抑制JNK激活以响应UV辐射的证据,表明LKB1在调节JNK应激信号响应于各种刺激中的基本作用。从进化的角度来看,我们推测LKB1抑制JNK信号的能力在面对能量或氧化还原应激的正常组织中可能是有利的,通过暂时抑制凋亡直到补偿机制 (也受LKB1调节) 可以订婚。尚不清楚JNK信号传导的调节是否有助于LKB1的肿瘤抑制功能,或者过度激活JNK信号传导的能力是否为失去LKB1的癌细胞提供了优势。值得注意的是,在MAPK抑制的设置中,最大限度地观察到由LKB1丢失赋予的差异JNK激活和MCL-1依赖性增加,这表明该途径的功能效应可能在特定环境中被掩盖,以响应选择的扰动。此外,我们观察到LKB1的敲低在不同的细胞系中不同程度地抑制了药物诱导的JNK激活,这表明其他上下文特异性途径可能协同调节JNK。
总之,我们确定了LKB1-NUAK调节JNK应激信号和调节凋亡依赖性的机制。KRAS-突变的非小细胞肺癌。响应于KRAS或MEK抑制,LKB1-deficient细胞表现出JNK的过度活化和增加的对MCL-1的依赖性以缓冲BIM的增加。虽然LKB1-deficiency不会增加对KRAS的敏感性G12C或MEK抑制剂用作单一药剂,当用MCL-1 BH3模拟物处理时,细胞变得为细胞凋亡引发。这些结果表明了一种潜在的生物标志物信息联合治疗方法,该方法基于突变或基因组丢失STK11/Lkb1。
方法细胞培养在我们的实验室中,使用先前描述的方法从恶性胸腔积液,芯针活检或手术切除中建立了患者来源的NSCLC细胞系 (PMID: 30254092),除了来源于原代小鼠PDX模型的MGH1070细胞系。所有患者均提供知情同意书,以参加dana-farber/哈佛癌症中心机构审查委员会批准的组织收集方案,并授予对其样品进行研究的许可。临床观察KRAS在已建立的细胞系中验证突变 (通过MGH快照NGS基因分型小组确定)。将建立的患者来源的细胞系维持在rpmi + 10% FBS中。公众可获得的NSCLC细胞系从麻省总医院癌症中心的分子治疗中心获得; STR验证在项目 (生物合成公司) 开始时进行。在实验使用期间常规测试细胞系的支原体。将细胞系维持在rpmi + 5% FBS中,除了A427、SW1573、H2009和H1573维持在DMEM/F12 +.5% FBS中。
细胞增殖评估使用celltiter-glo测定 (Promega) 评估细胞增殖。在加入药物前24小时将细胞接种到96孔板中。加入药物后72小时,通过将细胞与celltiter-glo试剂在室温下在振荡平台上孵育30分钟来测定细胞增殖。使用spectramaxi3x读板器 (Molecular Devices) 定量发光。
PI/膜联蛋白凋亡检测在加入药物前24小时以低密度接种细胞。加入药物后72小时,收集粘附 (活) 和漂浮 (死) 细胞,用碘化丙啶 (PI) 和Cy5-Annexin V (BD生物科学) 染色并通过流式细胞术分析。使用FlowJo软件对膜联蛋白阳性凋亡细胞分数进行定量。
工程细胞系的产生EV和LKB1细胞系空载体 (EV,pBabe) 和LKB1逆转录病毒载体是博士的礼物。kwok-kin Wong (纽约大学)。通过使用Lipofectamine 3000 (ThermoFisher) 用EV或LKB1,vsv-g (Addgene #8454),gag-pol (Addgene #14887) 转染HEK293T细胞来制备EV和LKB1病毒并收集上清液中的病毒颗粒。通过转导产生稳定的细胞系KRAS-具有EV或LKB1病毒的突变NSCLC系,随后进行嘌呤霉素选择。
LKB1敲除细胞系Sgrna靶向STK11使用chop-chop设计基因座并克隆到pSpCaS11(BB)-2A-GFP (Addgene #48138) 中。KRAS-用质粒瞬时转染突变的NSCLC细胞系,并通过FACs分选以形成单克隆。克隆扩增后,选择20个克隆,并通过western印迹评估LKB1表达的丧失。或者,将lkb1sgrna克隆到皮孔rispr v2 (Addgene #52961) 中。通过使用lipofectamine3000 (ThermoFisher) 用EV或sgLKB1、vsv-g (Addgene #8454) 和 Δ8.91转染HEK293细胞来制备慢病毒颗粒。通过感染产生稳定的细胞系KRAS-具有皮孔病毒v2或sgLKB1病毒的突变NSCLC系,然后进行嘌呤霉素选择。
DOX诱导MCL-1,bcl-xl细胞系合成全长野生型或突变MCL-1 bcl-xl编码序列 (GenScript) 并将其克隆到pInducer20 (来自leezou,MGH的gift) 中。通过用pInducer20或pInducer20-MCL-1/ pInducer20-BCL-XL、vsv-g (Addgene #8454) 和 Δ8.91使用lipofectamine3000 (ThermoFisher) 转染HEK293细胞来制备慢病毒颗粒。通过感染产生稳定的细胞系KRAS-用EV或pInducer20-MCL-1或pInducer20-BCL-XL病毒感染突变NSCLC系,然后用新霉素/g418选择。
小鼠异种移植研究KRAS-突变体通过将来自恶性胸腔积液的肿瘤细胞或组织皮下植入雄性NSG小鼠 (jacksonlabs) 、芯针活检或手术切除产生nsclcpdx模型。所有患者都签署了知情同意书,以参加Dana Farber/哈佛大学癌症中心机构审查委员会批准的方案,允许对其样本进行研究。根据机构指南,通过MGH机构动物护理和使用委员会 (IACUC) 批准的动物方案进行所有动物研究。将皮下肿瘤连续传代两次以完全建立每个模型。临床观察KRAS在每个建立的模型中验证突变。使用的所有小鼠在8周至6月龄之间。将小鼠保持在74 °F ± °2 °F (23 °C ± 1 °C) 的温度下,具有12小时光照/12小时黑暗循环。相对湿度保持在30-70%。每个实验中使用的小鼠编号显示在图例中。最大肿瘤大小不超过2000毫米3,符合IACUC规定。对于药物研究,将PDX肿瘤组织直接皮下植入NSG或无胸腺裸鼠 (Nu/Nu) 中。对于H2030异种移植物研究,在1:1基质胶: PBS和5 × 10中制备细胞系悬浮液6将细胞单侧注射到雌性裸 (Nu/Nu) 小鼠的侧腹上的皮下空间中。用电子卡尺测量肿瘤,根据公式v = 0.52 × l × w计算肿瘤体积2。具有既定肿瘤的小鼠 (250-400 °mm3) 使用协变量自适应随机化将其随机分配到药物治疗组,以最大程度地减少基线肿瘤体积的差异。将曲美替尼溶解在0.5% HPMC/0.2% tween80 (ph8.0) 中,并通过口服管饲法以3 °c/kg每日施用,每周6天。将Sotorasib溶解在2% HPMC/0.1% tween80 (ph7) 中,并通过口服管饲法以100 mg/kg每日施用,每周6天。将amg176溶解在25% 羟丙基 β-环糊精 (pH8.0) 中,并通过口服管饲法每天施用50 mg/kg。
定量rt-pcr分析使用qiagenrneasy试剂盒提取RNA。使用oligo-dt引物,用转录高保真cDNA合成试剂盒 (Roche) 制备cDNA。用基因特异性引物进行定量PCR (补充数据
2) 使用SYBR™在Lightcycler 480 (ThermoFisher) 上选择主混合物 (应用生物系统)。相对基因表达是通过使用 Δ Δ ct方法通过归一化来计算的
ACTB。
将细胞接种在6孔或6cm平板中,当细胞达到70% 汇合时加入药物。通过用PBS洗涤两次收获细胞,在裂解缓冲液中裂解
16在冰上,并在4 ℃ 下以14,000 rpm旋转10 ℃ 以除去不溶性细胞碎片。通过二辛可宁酸测定法 (ThermoFisher) 测定裂解物蛋白质浓度。使用NuPage 4-12% Bis-trismidi凝胶 (Invitrogen) 在nupagemopssds运行缓冲液 (Invitrogen) 中进行凝胶电泳,然后转移到PVDF膜 (ThermoFisher) 上。转移后,膜用Tween 20 (tbs-t) 的Tris缓冲盐水中的5% 牛奶 (Lab Scientific bioKEMIX) 封闭,然后与一抗 (1:1000,1% BSA的tbs-t溶液) 在4 °C过夜。在tbs-t中洗涤后,将膜与适当的第二抗体 (在tbs-t中的2% 脱脂乳中1:12500) 在室温下孵育1 °c。使用以下HRP连接的第二抗体: 抗兔igg (CST7074) 和抗小鼠IgG (CST7076)。从第二抗体中除去膜,并在tbs-t中洗涤3次,每次10分钟。在成像之前,将膜孵育4 °min SuperSignal West Femto稳定过氧化物和鲁米诺/增强剂 (thermofisher),在0.1 °mtris-HCL ph8.8 (波士顿生物产品公司) 中以1:10稀释。使用G: boxchemi-xrq系统 (Syngene) 对发光成像。使用以下一抗: pJNK T183/Y185 (CST4668),SAPK/JNK (CST9252),BIM (CST2933),pbcl-xl S112 (Invitrogen 44-428 G),bcl-xl (CST2764) 、LKB1 (CST3050) 、pMCL-1 T163 (CST14765) 、pMCL-1 S159/T163 (CST4579) 、pMCL-1 S114 (CST13297) 、MCL-1 (BD Pharmingen 559027) 、pMKK4 S257/T261 (CST9156),MKK4 (CST9152) 、pMEK7 S271 (赛默飞世尔PA5-114604) 、pMEK7 T275 (赛默飞世尔PA5-114605) 、MKK7 (CST4172) 、DUSP10/MKP5 (CST3483) 、HA标签 (CST3724) 、 Β-微管蛋白 (CST2146),GAPDH (CST5174)。将用于蛋白质印迹分析的所有抗体稀释至1:1000的浓度。图
3H: 样品来自相同的实验,但LKB1、pJNK和JNK的凝胶不同。样品来自相同的实验,但对于PP1B和GAPDH凝胶不同。图
3K: 样品来自相同的实验,但PP1B和I-2的凝胶不同。图
5B-E: 样品来自相同的实验,但BIM和bcl-xl的凝胶不同。无花果。
S5B: 样品来自相同的实验,但LKB1和pAMPK的凝胶不同。无花果。
S6A, D, H: 样品来自相同的实验,但LKB1、pJNK、JNK和GAPDH的凝胶不同。无花果。
S8A, B: 样品来自相同的实验,但用于pJNK、JNK和GAPDH的凝胶不同。无花果。
S8E, F: 样品来自相同的实验,但PP1B和GAPDH的凝胶不同。无花果。
S9F: 样品来自相同的实验,但LKB1、pAMPK和肌动蛋白的凝胶不同。图。
S10 A, B, E: 样品来自相同的实验,但BIM和bcl-xl的凝胶不同。图。
S11A, B: 样品来自相同的实验,但pMCL-1、pJNK和GAPDH的凝胶不同。图。
S11C: 样品来自相同的实验,但LKB1、pJNK、pMCL-1和GAPDH的凝胶不同。图S
11F: 样品来自相同的实验,但bcl-xl和HA的凝胶不同。图。
S11G: 样品来自相同的实验,但pMCL-1 S64、pMCL-1 S159和pMCL-1 t163的凝胶不同。图。
S11L: 样品来自相同的实验,但BIM和bcl-xl的凝胶不同。
将细胞接种在10厘米或15厘米平板中,当细胞达到70% 汇合时加入药物。在处理期后收获细胞,并使用tris裂解缓冲液与蛋白酶抑制剂混合物 (mesoscale Discovery) 在冰上制备裂解物。总蛋白浓度标准化后,Pierce蛋白A/G磁珠 (ThermoFisher) 和小鼠抗人MCL-1 (4 μ G/反应,BD Pharmingen 559027) 或将小鼠抗人bcl-xl (4 μ g/反应,emdmilliporemab3121) 抗体加入到裂解物等分试样中,并在4 ℃ 孵育过夜。保存标准化的全细胞裂解物的代表性等分试样用于蛋白质印迹分析。使用磁分离分离免疫沉淀的级分,用tris裂解缓冲液在冰上洗涤三次,通过用tris裂解缓冲液和LDS样品缓冲液4X (Invitrogen) 在95 °C加热10 °C洗脱蛋白质。对于蛋白质印迹,使用兔抗人MCL-1 (1:1000,CST4572) 抗体; 所有其他抗体与用于蛋白质印迹的那些相同。对于HA标记的bcl-xl的免疫沉淀,按照制造商的方案使用Pierce磁性HA标记IP/co-ip试剂盒 (thermofisher) (特别是,(A) 的程序手册IP/co-ip和 (B.)用于减少凝胶分析的洗脱方案2)。
免疫荧光和图像分析将细胞用10% 中性缓冲的福尔马林固定,并通过PBST (pbs + tritonx100) 透化。然后将细胞与pjnkt183/Y185 (CST4668) 一抗 (1:200) 在4 ℃ 孵育过夜。oC.第二抗体染色在室温下进行1 °c,然后进行DAPI染色。使用zeisslsm710共聚焦显微镜获取图像。使用CellProfiler软件 (Broad Institute) 进行图像分析。简言之,通过DAPI染色鉴定单个细胞。将细胞核内或细胞核外的pJNK染色分段并在单个细胞水平上定量。
免疫组织化学解剖异种移植肿瘤并在4% 多聚甲醛 (sigma158127) 中固定。通过MGH组织病理学核心进行石蜡包埋和切片。将组织载玻片在60 °C下烘烤10 °C,然后脱水。通过在压力锅中在基于柠檬酸盐的溶液 (vectorlab H-3300-250) 中孵育来进行抗原修复。然后将载玻片在3% 过氧化氢的甲醇溶液中孵育10分钟,并用蒸馏水洗涤两次。然后将载玻片在封闭溶液 (pJNK和pAMPK: tbs-t中的3% BSA,LKB1: cas-block thermofisher8120) 中孵育,然后用1稀释的第一抗体: 200在tbs-t (pJNK CST 4668s,pAMPK CST 2535s,LKB1 CST 3050s) 中于4 ℃ 过夜。在初级抗体孵育后,用tbs-t洗涤载玻片,并与次级生物素化抗体和vectaustaelitabc试剂 (vectorlab PK-6101) 一起孵育。使用DAB底物试剂盒 (vectorlab SK-4100) 进行HRP检测并用苏木精 (sigma51275) 复染。然后将载玻片脱水并固定。使用leicadm2500显微镜进行组织成像。Tbs-t: 含有吐温的Tris缓冲盐水。
siRNA介导的基因敲低根据制造商的方案 (Invitrogen,Cat #13778075),使用lipofectaminernaimax转染试剂进行siRNA转染。简言之,将细胞接种在6孔、6 °c或10 °c的板中,并且当细胞达到 ~ 70% 汇合时进行siRNA转染。在转染之前,将细胞置于无抗生素的培养基中。转染后48小时,接种细胞用于增殖分析或免疫沉淀,或收获细胞用于western印迹。使用以下invitrogensirna: 阴性对照 (NC) (AM4611) 、MAPK8 (ID: s11152) 、MAPK9 (ID: s11159) 、NUAK1 (ID: S110) 、NUAK2 (ID: s37779), PRKAA1 (ID: S120), PRKAA2 (ID: s11056), PRKAB1 (ID: s11059), PRKAB2(ID: s11062), SIK1 (ID: S115377), SIK2 (ID: s23355), SIK3 (ID: s23712), MARK1 (ID: S12511), MARK2 (ID: s11648) 、MARK3 (ID: S12514) 、MARK4 (ID: s33718) 、MAP2K4 (ID: s11182、s11183) 、MAP2K7 (ID:s11183,s11184),MCL-1 (ID: S12584,S12585),BCL2L1 (ID: s1920,s1921,s1922)。
细胞系的BH3谱如前所述,通过定量添加外源BH3肽后的细胞色素c释放来进行BH3谱分析
49。简单地说,2 × 106分离细胞,在500 × × ℃ 离心
g5 ℃ 分钟,然后将细胞沉淀重悬于具有1 ℃ 僵尸绿色活力染料 (BioLegend,cat #423111) 的100 ℃ μ lpbs中。将细胞在室温下在光线下染色15 ℃,然后通过添加400 ℃ 的FACS染色缓冲液 (PBS中的2% FBS) 淬灭。然后将细胞在500 × × ℃ 离心。
g持续5 ℃ 分钟,并使用指示的肽和浓度进行BH3谱分析。在BH3分析后,将细胞用基于皂苷的缓冲液 (1% 皂苷、10% BSA的PBS溶液) 透化用于细胞内染色。并用细胞色素c AlexaFluor 647抗体 (BioLegend 612310; 1:2000稀释) 和DAPI在4 °C染色过夜。次日用流式细胞术 (Attune NxT) 分析细胞。
所有患者都签署了知情同意书,以参加Dana Farber/哈佛大学癌症中心机构审查委员会批准的方案,允许对其样本进行研究。患者年龄在44-78岁之间,其中6名 (67%) 女性和3名 (33%) 男性。手术切除将肿瘤组织手动切碎至 ~ 1 mm3并在不存在或存在药物的情况下在RPMI1640 °c 10% FBS中孵育过夜。在BH3谱分析之前,通过胶原酶/分散酶在37 °C下酶解30分钟进一步解离组织。然后将样品通过100 μ m过滤器过滤以分离单细胞。对于每个样品,2 × 106分离细胞,在500 × × ℃ 离心
g持续5 °min,并将细胞沉淀重悬于具有1 °yl僵尸绿色活力染料 (BioLegend,cat #423111) 的100 °yl PBS中。将细胞在室温下避光染色15 ℃,然后通过添加400 ℃ 的FACS染色缓冲液 (PBS中的2% FBS) 淬灭。将细胞在500 × × ℃ 离心
g5 °min,并重悬于100 °μlfacs染色缓冲液中。然后用以下缀合的细胞表面标志物抗体以1:50稀释度染色细胞: epcampe (BioLegend,324206) 和cd45bv786 (BioLegend,304048)。然后将细胞在500 × × ℃ 离心。
g持续5 °min,并如前所述进行BH3分析
49用指定的肽 (例如,MS1 = MCL-1,hrk = bcl-xl) 和浓度。BH3分析后,将细胞用基于皂苷的缓冲液 (1% 皂苷,10% BSA在PBS中) 透化以进行细胞内染色,并用细胞色素c AlexaFluor 647的抗体染色 (BioLegend,612310; 1:2000稀释) 和DAPI。将细胞在4 °C染色过夜,并在第二天通过流式细胞术 (attunenxt) 进行分析。鉴定了感兴趣的细胞: DAPI阳性、僵尸阴性、CD45阴性和EpCAM阳性。
所有磷酸-蛋白质组学样品作为单个生物重复进行分析。将细胞沉淀冷冻,裂解,用DTT还原,然后用碘乙酰胺烷基化。按照MeOH/CHCl3方案沉淀蛋白质,用胰蛋白酶和LysC消化。如前所述进行磷肽富集
39,
82,
83。对于每个样品,用TiO2珠 (glsciences,日本) 从2.5mg肽进行磷酸肽富集。用TMT10plex试剂 (thermofisherscientific) 进行磷酸肽标记,然后将样品合并并通过碱性pH反相色谱法分离成24个级分,如前所述
84。将分级分离的肽干燥,溶解在5% ACN/5% 甲酸中,并使用同时前体选择 (SPS) 支持的MS3方法在Orbitrap FusionLumos质谱仪上通过3-h LC-M2/MS3运行进行分析
85,
86如前所述
87。对于每种肽,使用CID和HCD片段化获得两个MS2光谱
88。使用基于SEQUEST的定制蛋白质组学分析流程分配每个MS2光谱
89允许酪氨酸、苏氨酸和丝氨酸残基的磷酸化作为可变修饰。使用Ascore算法评估肽序列内磷酸化的正确分配
90。使用目标-诱饵数据库搜索策略将蛋白质和肽分配过滤至错误发现率 (FDR) <1%
91并采用线性判别分析和后验误差直方图排序
89。将UniProt数据库 (2014) 中与一种以上蛋白质相对应的肽序列分配给匹配肽数量最多的蛋白质
89。对于MS3光谱,只有那些具有平均信噪比 > 40每个报告离子和分离特异性的光谱
86 > 0.75被量化。通过两步法将蛋白质TMT强度归一化。首先根据所有蛋白质的中值平均蛋白质强度在每种蛋白质的所有TMT通道上对P-蛋白强度进行归一化。通过计算来自每个TMT通道中所有蛋白质的归一化强度的中值来校正肽混合物中每个样品的轻微混合误差,然后将各个蛋白质强度归一化为中值强度值。
使用TMT-10plex试剂 (thermoscientific) 标记来自上述胰蛋白酶消化物的肽 (50 μ g)。合并后,通过碱性反相HPLC分离标记的样品
84。通过在Orbitrap FusionLumos上运行3小时反相LC-MS2/MS3来分析馏分。使用SEQUEST算法搜索整个人类数据库 (uniprot 2014) 进行来自MS2光谱的蛋白质鉴定
92使用自定义分析管道
89。搜索策略包括基于目标诱饵数据库的搜索,以过滤错误发现率 (FDR) <1%
91。用于定量的MS3分离使用如前所述的同时前体选择 (SPS)
85,
86,
87。只有MS3的每个报告离子的信噪比> 40,并且具有分离特异性
86> 0.75; 如上所述进行使用两步归一化过程的归一化。
使用PTM特征富集分析 (pmt-sea) 进行磷酸化特征分析,这是ssGSEA2.0的修改版本 (
https://github.com/broadinstitute/ ssGSEA2.0)。简言之,通过比较每组中磷酸肽的水平来计算相对对数倍增加/减少。将相对对数倍增加/减少导入到pmt-sea包中,并与PTM签名数据库 (PTMsigDB) 进行比较。输出显著的签名,排列并在组之间比较 (例如LKB1-positive对LKB1-negative等基因对)。
使用生物化学直观的广义Loewe (BIGL) 进行协同分析
93。根据增加剂量的曲美替尼/索托拉西布和amg176的2 °x °2矩阵处理NSCLC细胞系72小时,并通过celltiter-glo评估细胞活力。协同作用分析分为三个步骤: 1.使用非线性最小二乘估计确定每种化合物的边际曲线。2.“一般Loewe模型” 的预期效果是根据先前计算的边际曲线计算得出的。3.使用maxR统计检验将预期响应与观察到的可行性进行比较,该检验评估空模型是否局部拟合观察到的数据。
使用student进行所有实验的统计测试t测试,单向或双向方差分析。对于学生t测试,配对的、未配对的、参数的或参数的测试对于如在相应的图例中指定的个体实验进行。所有测试均使用双侧假设进行。双向ANOVA用于具有Tukey校正的组的多重比较 (95% 置信区间)。蛋白质印迹和免疫沉淀实验显示的数据代表至少两个独立的生物学重复。免疫荧光和免疫组织化学实验的图像代表至少3个生物学重复。没有使用统计方法来预先确定样本量。没有数据被排除在分析之外。在实验和结果评估期间,研究人员对分配没有不知情。
报告摘要
数据可用性








 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡